Clinical-molecular profiling of atypical GNAO1 patients: Novel pathogenic variants, unusual manifestations, and severe molecular dysfunction
Clinical-molecular profiling of atypical GNAO1 patients: Novel pathogenic variants, unusual manifestations, and severe molecular dysfunction
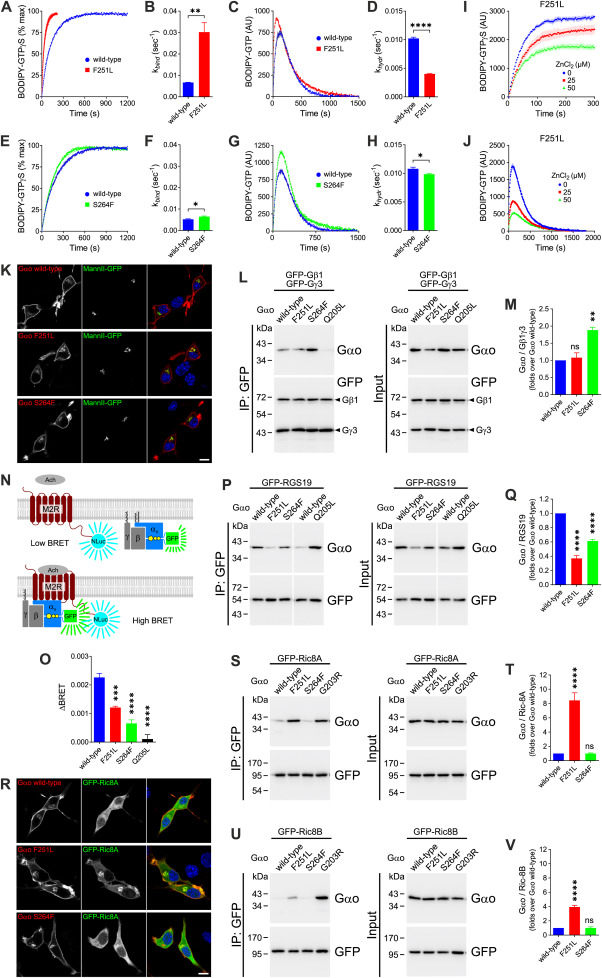
G protein subunit alpha O1 (GNAO1)-related disorders represent a broad spectrum of neurological diseases mainly caused by de novo mutations in GNAO1 encoding for G protein alpha subunit o (Gαo). As the major transducer of neuronal G protein-coupled receptors (GPCRs), Gαo is essential for the signaling involved in neuronal excitability and neurodevelopment. The most severe neomorphic GNAO1-mutations lead to developmental and epileptic encephalopathy-17 (DEE17; OMIM #615473) or neurodevelopmental disorder with involuntary movements (NEDIM; OMIM #617493), the latter with or without epileptic seizures.1 Movement disorders are present in almost all patients, with hypo/hyperkinetic features and profound impairment of postural development.2 Milder phenotypes including late-onset dystonia and parkinsonism with different extents of cognitive impairment have recently emerged from mutations leading to loss-of-function and haploinsufficiency.3 However, clear genotype–phenotype correlations and underlying pathogenic mechanisms are still poorly understood, limiting an accurate prediction of disease progression and the implementation of early therapeutic interventions. Here, we present two unrelated Italian patients carrying novel GNAO1 mutations with atypical phenotypes, thus expanding the phenotypic spectrum of GNAO1-related disorders. We additionally provide a deep molecular analysis of the pathogenic Gαo variants along with a discussion of potential treatment options.