The mucolipidosis III-causing mutation in GNPTAB, c.1760G>C, disrupts the development of somites in rats
The mucolipidosis III-causing mutation in GNPTAB, c.1760G>C, disrupts the development of somites in rats
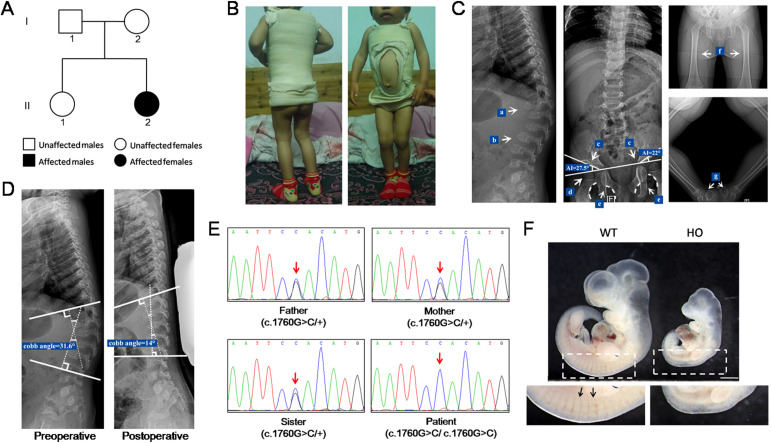
Mucolipidosis (ML) II (OMIM 252500) and III α/β (OMIM 252600) are a group of rare lysosomal storage disorders caused by mis-sorting of lysosomal hydrolases and the subsequent accumulation of nondegraded macromolecules. These disorders manifest as multiple-systemic abnormalities throughout the body, mainly including severe dysplasia, short stature, scoliosis, joint stiffness, joint contractures, claw-hand deformities, craniofacial deformities, retinal degeneration, mental retardation, cognitive impairment, and internal organ dysfunction. ML II has an early onset, and patients usually die in early childhood. For ML III α/β, symptoms are less severe due to residual lysosomal enzymes, with a late onset and slow progression; these patients usually die in adulthood.1 Both ML II and ML III α/β are inherited in an autosomal recessive manner and caused by mutations in GNPTAB.