Rare variants in NRSN2 cause non-syndromic orofacial cleft through dysregulation of TGF-β signaling
Rare variants in NRSN2 cause non-syndromic orofacial cleft through dysregulation of TGF-β signaling
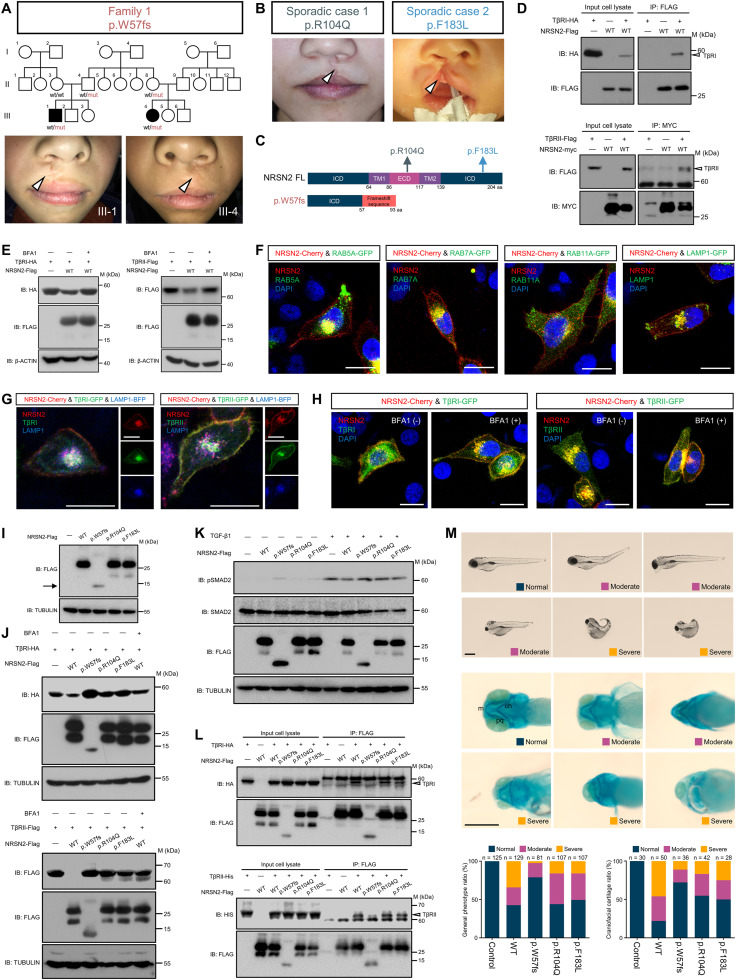
Orofacial cleft (OFC) is the most common congenital craniofacial disorder that significantly affects the appearance and orofacial function of patients. Although previous studies have demonstrated that rare variants are significant contributors to the genetic etiology of non-syndromic OFC (NSOFC),1 only in a limited number of cases the underlying causal genes are identified. The human Neurensin 2 (NRSN2) gene, encoding a two-transmembrane-domain protein,2 has not previously been associated with craniofacial development or malformations. In this study, we performed whole-exome sequencing (WES) and target-region sequencing (TRS) on 10 multiplex families and 138 sporadic cases with NSOFC, identifying three rare variants in the NRSN2 gene. Functional analyses in mammalian cells revealed that NRSN2 interacts with and degrades type 1 and type 2 TGF-β receptors (TβRI and TβRII) through the endosome–lysosome pathway, thereby inhibiting TGF-β signaling. In contrast, all three NRSN2 variants show an impaired capacity to degrade TβRI and TβRII, causing unrestrained TGF-β signaling. Consistently, these NRSN2 variants are less effective to induce craniofacial abnormalities in zebrafish embryos, corroborating that they are loss-of-function (LoF) mutations. Taken together, we conclude that NRSN2 variants behave as hypomorphic alleles that result in aberrant TGF-β signaling and thus NRSN2 is a novel causal gene for NSOFC in humans.