Novel PIK3CG compound heterozygous variants cause inactivated PI3Kγ syndrome presenting as necrotizing enterocolitis in a preterm infant
Novel PIK3CG compound heterozygous variants cause inactivated PI3Kγ syndrome presenting as necrotizing enterocolitis in a preterm infant
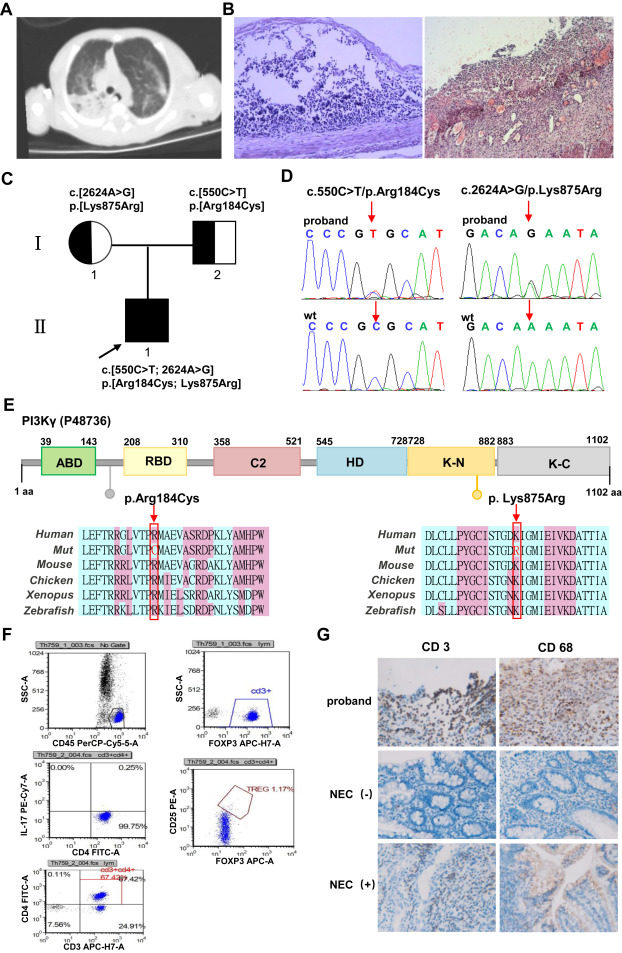
Inactivated phosphoinositide 3-kinase gamma (PI3Kγ) syndrome (IPGS; OMIM #619802), an autosomal recessive immunologic disorder first described by Takeda et al in 2019, classically manifests in childhood with recurrent infections, pneumonia, and colitis.1 This disorder is caused by biallelic loss-of-function variants in the PIK3CG (OMIM ∗601,232), located at 7q22.3, encoding the catalytic subunit p110γ of the PI3Kγ enzyme. The p110γ subunit, predominately expressed in immune cells and responsible for chemotaxis, reactive oxygen species (ROS) generation, and cytokine generation, also maintains critical roles in endothelial cells, neurons, cardiomyocytes, and lung cells.2,3 Pathogenic variants in PIK3CG disrupt PI3K signaling, leading to immune dysregulation characterized by antibody deficiency, excessive T cell infiltration in the lungs/intestines, and significantly disrupted levels of T regulatory cells.1,4