Novel heterozygous missense variants in the TOE1 gene linked to pontocerebellar hypoplasia type 7
Novel heterozygous missense variants in the TOE1 gene linked to pontocerebellar hypoplasia type 7
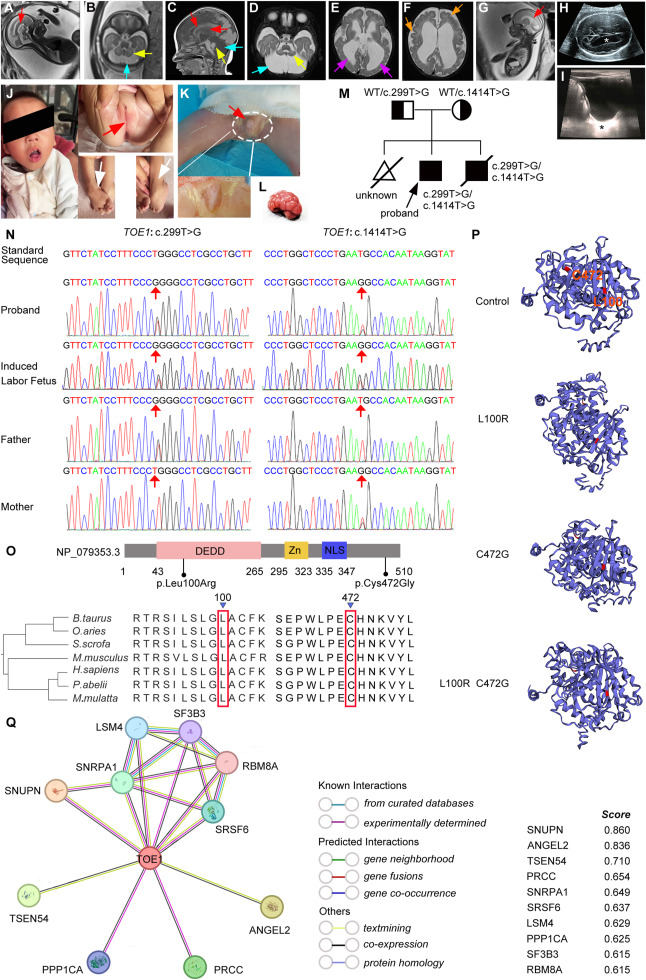
Pontocerebellar hypoplasia type 7 (PCH7) (OMIM #614969) stands as a rare and severe neurodegenerative syndrome marked by distinct characteristics, including neurological decline, hypoplasia in the pons and cerebellum, muscle hypotonia, irregular breathing, and hypogonadism.1 Furthermore, individuals with 46, XY karyotypes display feminine genitalia, whereas those with 46, XX karyotypes exhibit atrophic ovaries and lack menarche in PCH7 patients.2 Recent investigations have pinpointed variants in the EGR1 protein 1 gene (TOE1) as the genetic culprits behind PCH7 onset.3 TOE1 primarily situated within the Cajal bodies of the nucleus has been identified. In this cellular compartment, TOE1 functions as a 3-exonuclease, aiding in the maturation of small nuclear RNAs (snRNAs) and the processing of snRNA 3′-tails.4 Disruptions in snRNA processing may contribute to severe neurodegenerative disorders.