A novel homozygous missense variant in ARSK causes MPS X, a new subtype of mucopolysaccharidosis
A novel homozygous missense variant in ARSK causes MPS X, a new subtype of mucopolysaccharidosis
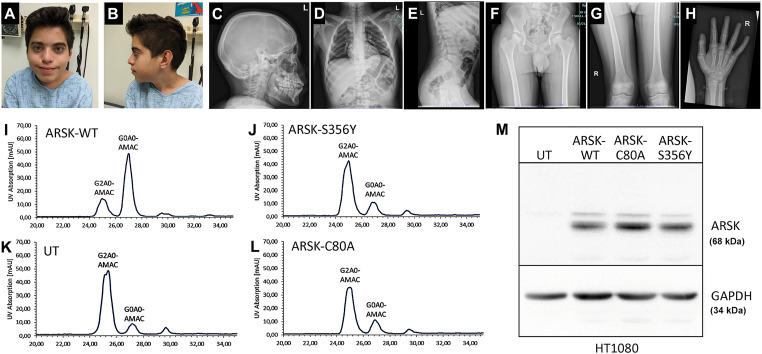
Mucopolysaccharidoses (MPS) are a group of rare inborn errors of metabolism caused by defective lysosomal enzymes which prevent cells from degrading and recycling certain carbohydrates and fats, resulting in the storage of glycosaminoglycans in cells throughout the body. This leads to multisystem abnormalities involving bone, connective tissues, brain, blood, spinal cord, skin, and other tissues. In humans, seven distinct types and many subgroups of MPS have been classified, and each is linked to deficiencies of the specific enzymes. Very recently, two studies1,2 have suggested defects in ARSK may lead to a new MPS subtype, MPS X, in humans. Here we report a 13-year-old cognitively intact boy diagnosed with Perthes disease and pectus carinatum referred for possible skeletal dysplasia. Exome sequencing analysis of his peripheral blood sample revealed a novel homozygous missense variant, c.1067C>A (p.S356Y) in the ARSK gene, functionally proven to result in ARSK deficiency, evidenced by failure to remove the 2-O-sulfate group from 2-sulfoglucuronate. Urine glycosaminoglycan (GAG) examination with a more sensitive liquid chromatography-tandem mass spectrometry (LC-MS/MS) method confirmed a significant increase in dermatan sulfate, suggesting malfunction of glycosaminoglycan metabolism. Skeletal abnormalities of vertical striae of distal femoral bones and bilateral Perthes were consistent with those observed in previous cases.1,2 Based upon the collective results of functional studies, urine GAG analysis, and skeletal findings, the novel homozygous missense germline variant in ARSK (c.1067C>A, p.S356Y) was determined to cause an MPS disorder due to ARSK deficiency. This novel ARSK pathogenic variant will be useful in diagnosis/prognosis and highlights the important and unique roles of ARSK in the degradation of sulfated glycosaminoglycans and maintenance of normal lysosomal functions in humans.