Characterization of the molecular dysfunctions occurring in Aicardi-Goutières syndrome patients with mutations in ADAR1
Characterization of the molecular dysfunctions occurring in Aicardi-Goutières syndrome patients with mutations in ADAR1
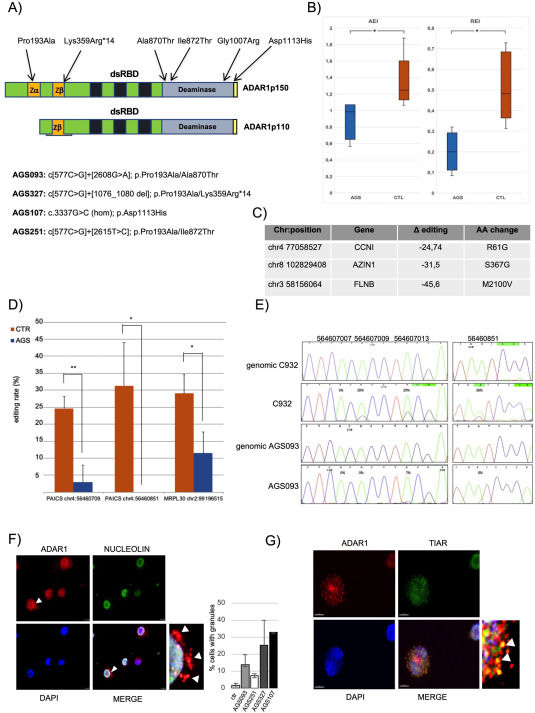
Aicardi-Goutières syndrome (AGS) is a systemic inflammatory disorder caused by mutations in any one of the nine different genes, whose deficiency provokes a type I (interferon) IFN response probably central to pathogenesis.1 ADAR1, one of the genes mutated in AGS (AGS6), encodes for an enzyme that belongs to the ADAR family (ADAR1, ADAR2, and ADAR3) that catalyzes the conversion of adenosine to inosine within double-stranded RNAs (dsRNAs) (RNA editing A-to-I).2,3 Two main isoforms of ADAR1 are expressed in mammals: the full-length p150 that is interferon-inducible and the constitutively expressed shorter isoform p110.2,3