[18F]florbetapir PET for early detection of amyloidosis in patients with hereditary transthyretin amyloidosis polyneuropathy
[18F]florbetapir PET for early detection of amyloidosis in patients with hereditary transthyretin amyloidosis polyneuropathy
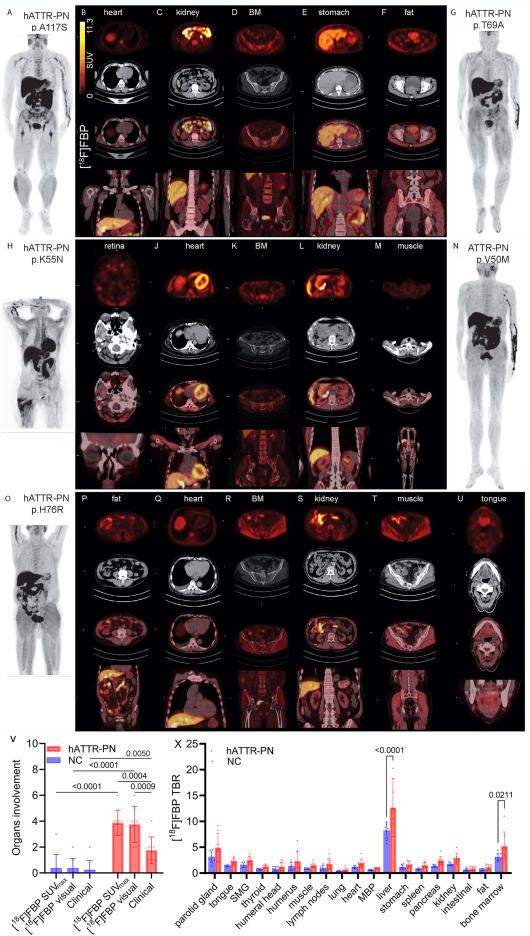
Systemic amyloidosis is a heterogeneous group of diseases characterized by localized or systemic deposition of insoluble extracellular fibrillary proteins in organs. Systemic amyloidosis is categorized by precursor protein, and the most common are immunoglobulin light-chain amyloidosis (AL) and transthyretin (TTR) protein produced predominantly in the liver (ATTR amyloidosis). ATTR amyloidosis frequently results from age-related misfolding of wild-type TTR and, less commonly, from misfolding of a variant TTR from an autosomal dominant mutation of the TTR gene.1 Hereditary ATTR (hATTR) is a progressive and potentially fatal disease with a heterogeneous clinical presentation; patients often develop a mixed phenotype of polyneuropathy (PN) characterized by sensory, motor, and autonomic neuropathy and/or cardiac amyloidosis.1 The survival rate varies widely, ranging from 3 to 15 years after diagnosis, depending on factors such as genetic mutation and clinical phenotype. It is extremely critical for the detection of amyloidosis at the earliest stage, when therapy is still effective before severe organ damage occurs.