Restored phagocytic ability of RPE patches derived from gene-corrected retinitis pigmentosa-hiPSCs on a biodegradable scaffold via clinical-grade protocol: Implications for autologous therapy
Restored phagocytic ability of RPE patches derived from gene-corrected retinitis pigmentosa-hiPSCs on a biodegradable scaffold via clinical-grade protocol: Implications for autologous therapy
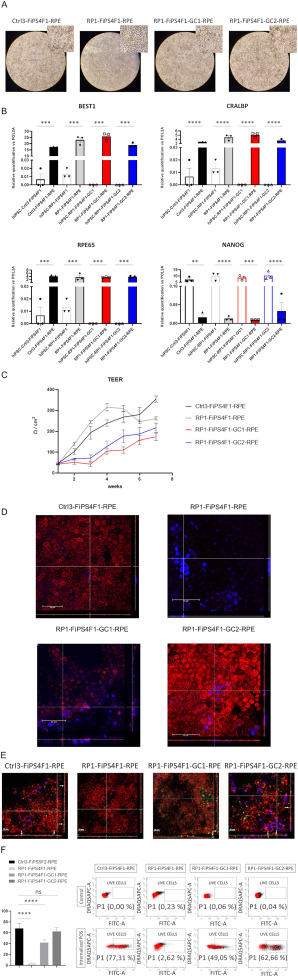
Cases of inherited retinal dystrophy (IRD) can be caused by mutations in the MERTK gene, which results in an autosomal recessive form of blindness (retinitis pigmentosa, RP) characterized by impaired phagocytosis of photoreceptor outer segments (POS) by retinal pigment epithelial cells (RPE). Persistent MERTK gene mutations in patient-derived human induced pluripotent stem cells (hiPSCs) pose a challenge for autologous stem cell-derived RPE replacement therapies targeting IRD. In a previous study, we created a hiPSC-based disease model of early-onset RP caused by a mutation in the human MERTK gene (homozygous frameshift mutation c.992_993delCA(p.Ser331Cysfs∗5)).1 The hiPSC patient's derived RPE showed impaired POS phagocytosis.1 Applying CRISPR/Cas9 gene-editing technology supported the generation of heterozygously and homozygously corrected RP-hiPSC lines, RP1-FiPS4F1-GC1 and RP1-FiPS4F1-GC2, respectively.2 The RPE cells derived from these isogenic hiPSC recovered both wild-type MERTK protein expression and recovered the function of phagocytosis of POS.