Dystrophin protein and mRNA analyses for the molecular genetic diagnosis of dystrophinopathy: A novel deep intronic DMD variant
Dystrophin protein and mRNA analyses for the molecular genetic diagnosis of dystrophinopathy: A novel deep intronic DMD variant
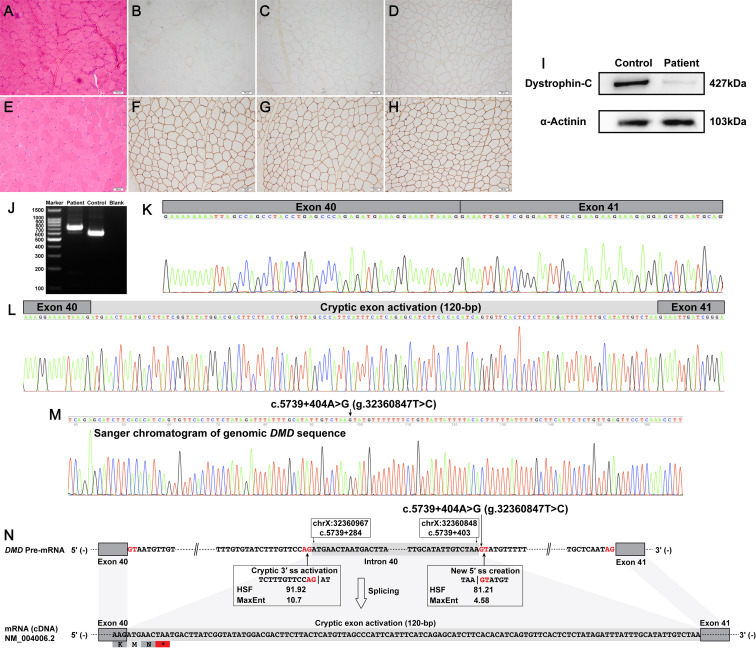
Becker muscular dystrophy (BMD) and Duchenne muscular dystrophy (DMD) are X-linked recessive muscular dystrophies caused by pathogenic dystrophin (DMD) variants.1 The prevalence of DMD and BMD in Caucasian communities was estimated to be approximately 4∼6 per 100 000 people, which was similar to the prevalence in Asian communities.2 Deletions and/or duplications of one or more canonical exons account for ∼80% of disease-causing variants in DMD. Most of the remaining ∼20% pathogenic dystrophin variants are subexonic small variants, which include small insertions and/or deletions, missense variants, nonsense variants, and canonical splice site variants. Multiplex ligation-dependent probe amplification analysis combined with genomic sequencing of all DMD canonical exons and flanking intronic sequences (referred to as the routine DNA-based techniques) can identify most exonic deletions, exonic duplications, and subexonic small variants that occur in DMD canonical exons and/or adjacent intronic sequences.1 Some rare and atypical pathogenic dystrophin variants can escape the detection of routine DNA-based techniques, which mainly consist of complex chromosomal rearrangements and deep intronic splicing-altering variants.1 Cryptic exon-activating variants are the most common type among deep intronic splicing-altering variants in DMD.1 Here, a novel deep intronic cryptic exon-activating variant in the human dystrophin gene (NM_004006.2:c.5739 + 404A > G) was identified by skeletal muscle tissue-derived dystrophin protein and mRNA analyses, genomic Sanger sequencing, and long-read whole DMD gene sequencing.