In vivo consequences of varying degrees of OTOA alteration elucidated using knock-in mouse models and pseudogene contamination-free long-read sequencing
In vivo consequences of varying degrees of OTOA alteration elucidated using knock-in mouse models and pseudogene contamination-free long-read sequencing
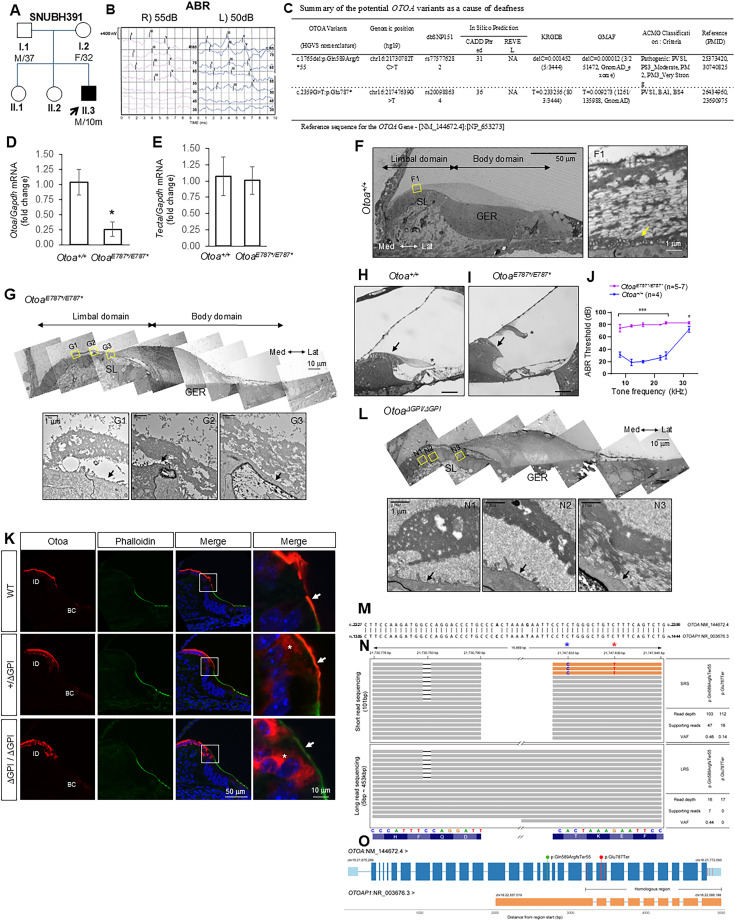
Otoancorin (OTOA) is a glycosylphosphatidylinositol (GPI)-anchored protein mediating the attachment of the tectorial membrane (TM) to the spiral limbus (SL) in the inner ear. Homozygous or compound heterozygous mutations in OTOA cause autosomal recessive deafness (DFNB22). We performed short-read exome sequencing (SRS) in a 10-month-old boy with sensorineural hearing loss, identifying a potential p.Glu787∗ variant in OTOA. Interestingly, this variant is common among normal-hearing individuals, leading us to question its pathogenic potential. We generated a knock-in mouse model for this variant and another lacking the C-terminal GPI-anchorage to study the in vivo consequences of deleting the C-terminus of Otoa. OtoaE787∗/E787∗ mice exhibited reduced transcript expression, TM detachment, and sensorineural hearing loss. Removal of the GPI-anchorage resulted in the loss of surface expression of Otoa and TM detachment, highlighting the importance of the C-terminus. To explain the discrepancy between the pathogenicity of p.Glu787∗ in the mouse model and its high allelic frequency in normal-hearing humans, we performed long-read sequencing (LRS) and identified that the variant was in a pseudogene (OTOAP1). Whole-genome sequencing revealed an inversion encompassing the 3′ end of OTOA in the patient. In summary, we demonstrated the limitations of SRS and confirmed the essential role of the Otoa C-terminus.