Properdin deficiency associated with systemic meningococcal disease due to a novel p.Cys337Arg pathogenic variant
Properdin deficiency associated with systemic meningococcal disease due to a novel p.Cys337Arg pathogenic variant
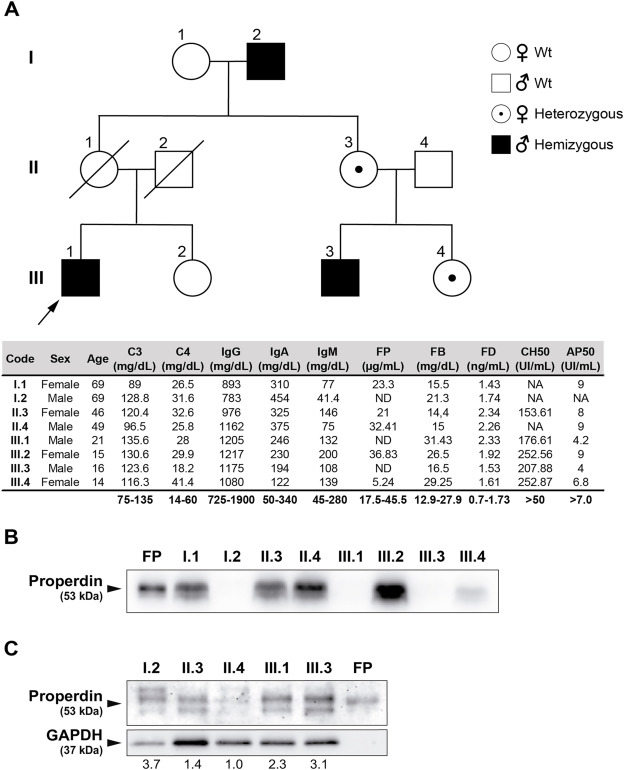
Properdin (FP) is a soluble glycoprotein with a key role in the regulation of the alternative pathway (AP) of the complement system.1 Neutrophils are the main source of FP, although monocytes, bone marrow progenitors, and T cells also synthesize this complement regulator. FP is codified by the gene CFP, located at Xp11.23–p11.3, and formed by 10 exons, of which exons 2 to 10 are translated conforming six thrombospondin type I repeats (TSR1-6).1 FP deficiency (MIM # 312060) is a rare X-linked disorder that contributes to increased susceptibility to infectious diseases, mainly caused by rare strains of Neisseria meningitidis such as serogroups W-135 and Y. Since its initial description in 1982,2 more than 100 cases have been reported worldwide demonstrating susceptibility to meningococcal infections and sepsis.1 FP deficiency can be divided into three subtypes: total deficiency (type I), partial deficiency (type II), and deficiency due to a dysfunctional protein (type III). In the present study, we investigated the members of a non-consanguineous Spanish family with a novel CFP mutation causing type I FP deficiency.