The 21-base pair deletion mutant Calpain3 does not inhibit wild-type Calpain3 activity
The 21-base pair deletion mutant Calpain3 does not inhibit wild-type Calpain3 activity
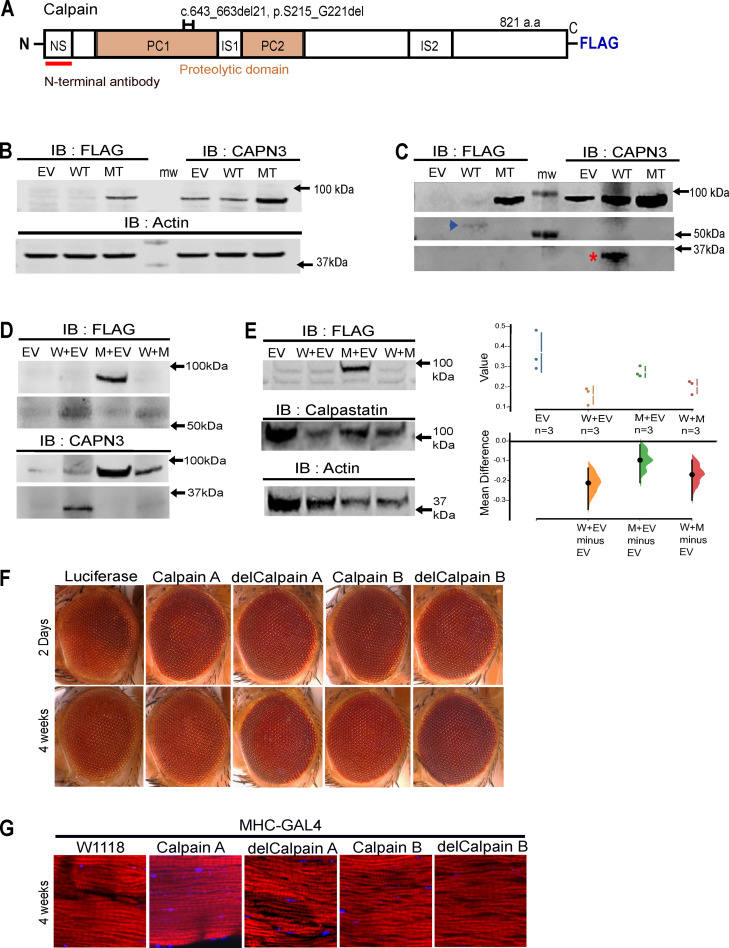
Calpain3 is one of the calpain protease family members that are predominantly expressed in skeletal muscle. Loss-of-function mutations in Calpain3 have been related to autosomal recessive limb-girdle muscular dystrophy 1 (LGMDR1), a common form of muscular dystrophy. Recently, the heterozygous 21-bp deletion mutation of the Calpain3 gene has been reported to cause autosomal dominant limb-girdle muscular dystrophy 4 (LGMDD4) which suggests that the mutant proteins act in a dominant-negative manner. Therefore, we examined whether the mutant protein could suppress the activity of wild-type Calpain3 using a cell culture model and a Drosophila model. The mutant Calpain3 resulted in catalytic inactivation, which did not inhibit wild-type Calpain3 autolytic and catalytic activities in HeLa cells. Overexpression of wild-type and mutant Calpain3 in Drosophila's eyes and muscles did not exhibit significant dominant toxicity. We provide evidence that mutant Calpain3 does not suppress wild-type Calpain3 activity. Rather, it is a mutant lacking autocatalytic activity like many other loss-of-function Calpain3 mutants causing LGMDR1. Our results implicate that the stability of the heteromeric mutant and wild-type Calpain3 complexes may be affected without inhibiting the wild-type activity per se. However, a thorough investigation is necessary to understand the molecular mechanism and dominant inheritance of the heterozygous 21-bp deletion mutation in LGMDD4.