A novel mutation in the KLHL17 gene is associated with neurodevelopmental disorders
A novel mutation in the KLHL17 gene is associated with neurodevelopmental disorders
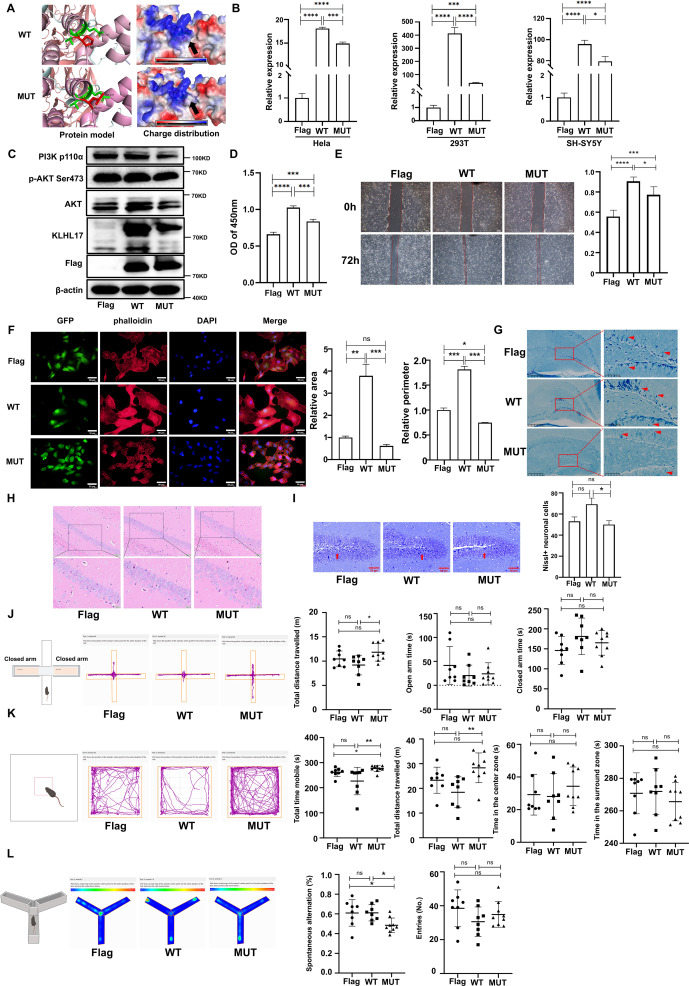
Kelch-like family member 17 (KLHL17) is predominantly expressed in the brain and plays a crucial role in neuronal development and function, deletions and/or mutations in KLHL17 have been linked to neurodevelopmental disorders in humans, e.g., intellectual disability, autism spectrum disorder, and infantile spasms, but the etiology and pathogenesis remain largely enigmatic.1,2 As a member of the family of the Kelch proteins, KLHL17 contains an N-terminal BTB/POZ domain followed by a BACK domain and four to six tandem Kelch motifs at the C-terminal region (Fig. S1A).1,3 Previously, we identified a novel de novo variant in KLHL17 (c.701C > T; p. P234L) in a cohort of 225 Chinese children with developmental delay/intellectual disability based on whole-exome sequencing (1/225), the mutation located in the BACK domain, a very high conversed region (Fig. S1B), and the affected boy presented with developmental delay, intellectual disability, hypotonia, and abnormal brainstem auditory evoked potential signal.4 The finding may offer a new clue to investigate the molecular pathogenesis of KLHL17 gene in neurodevelopmental disorders.