The genomic and epigenomic landscape of iridocorneal endothelial syndrome
The genomic and epigenomic landscape of iridocorneal endothelial syndrome
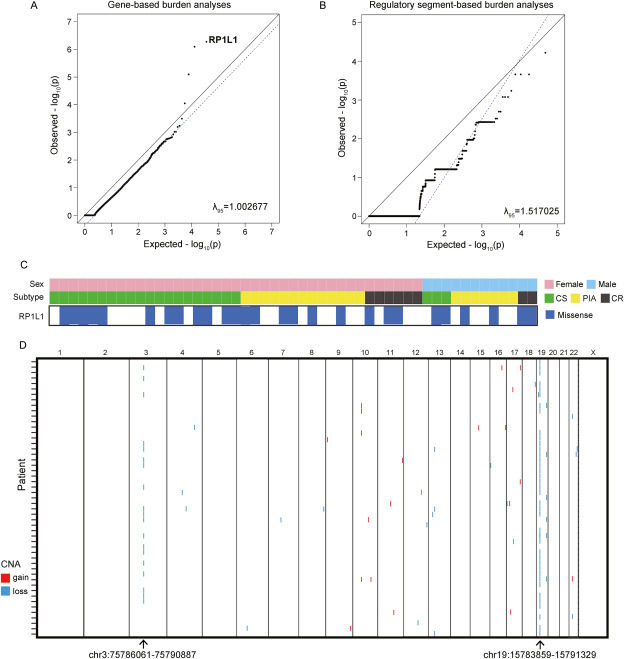
Iridocorneal endothelial (ICE) syndrome is a rare, irreversibly blinding eye disease with an unknown etiology. Understanding its genomic and epigenomic landscape could aid in developing etiology-based therapies. In this study, we recruited 99 ICE patients and performed whole-genome sequencing (WGS) on 51 and genome-wide DNA methylation profiling on 48 of them. We conducted mutational burden testing on genes and noncoding regulatory regions, comparing the ICE cohort with control groups (9197 East Asians from the gnomAD database and 350 normal Chinese from our in-house cohort). Copy number variation (CNV) analysis and differential methylation of regions were also explored. We identified RP1L1 (27/51, 53%) with a significantly higher coding-altering mutational burden in the ICE cohort (p < 8.3×10−7), with mutations predominantly at chr8:10467637 (hg19). Additionally, 41 regions with significant CNVs were identified, including two regions at chr19:15783859-15791329 (hg19) and chr3:75786061-75790887 (hg19), showing copy number loss in 39 and 19 patients, respectively. We also identified 2,717 differentially methylated regions (DMRs), with hypomethylation prevalent in ICE syndrome (91.9% of DMRs). Among these, 45 recurrent hypomethylated regions (HMRs) in more than 10% of ICE patients showed differential methylation compared to normal controls. This study presents the first comprehensive genomic and epigenomic characterization of ICE syndrome, offering insights into its underlying etiology.