Arhgap29 deficiency causes EEC like syndrome in mice
Arhgap29 deficiency causes EEC like syndrome in mice
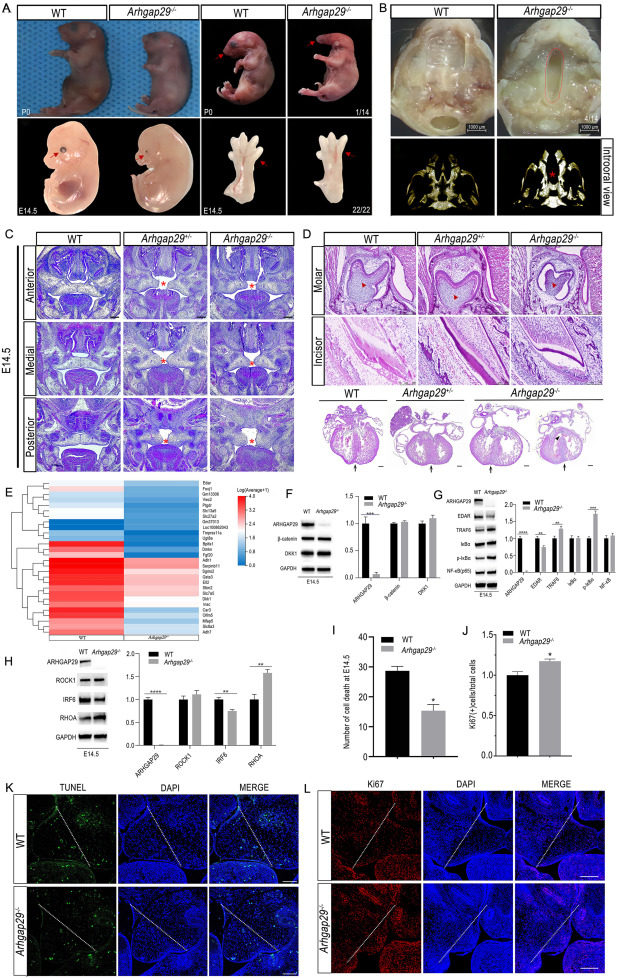
Cleft lip/palate (CL/P) is generally divided into two main types, non-syndromic and syndromic CL/P, with a global incidence of approximately 1 in 700.1 Previous studies have identified various susceptibility genes associated with non-syndromic CL/P, while the pathogenic gene of syndromic CL/P and the related mechanisms are not well documented. Ectrodactyly–ectodermal dysplasia–cleft lip/palate (EEC) syndrome is a representative syndromic CL/P, which is characterized by mild-to-severe symptoms such as missing or irregular fingers or toes, cleft lip or palate, distinctive facial features, and abnormalities of the eyes and urinary tract. The mutations in TP63 have been associated with some EEC cases. Meanwhile, there were other reported EEC cases without mutations in TP63 or other certain genes, suggesting other potential pathogenic genes involved in EEC syndrome. ARHGAP29 encodes Rho GTPase-activating protein 29, broadly expressed in various tissues such as blood, liver, and heart, especially in the palatal shelf.2, 3, 4 Arhgap29 mutant mice presented abnormal oral epithelial adhesions,5 ARHGAP29 has also been regarded as the susceptible gene of non-syndromic CL/P since 2012. Our whole exome sequencing also identified a new missense mutation(c.451C>T, p.Leu151Phe) in ARHGAP29 in a Chinese non-syndromic CL/P pedigree (Fig. S2C).