Profiling the cell diversity and tissue structure of aqueous humor circulatory system in human eyes using spatial single-cell RNA sequencing
Profiling the cell diversity and tissue structure of aqueous humor circulatory system in human eyes using spatial single-cell RNA sequencing
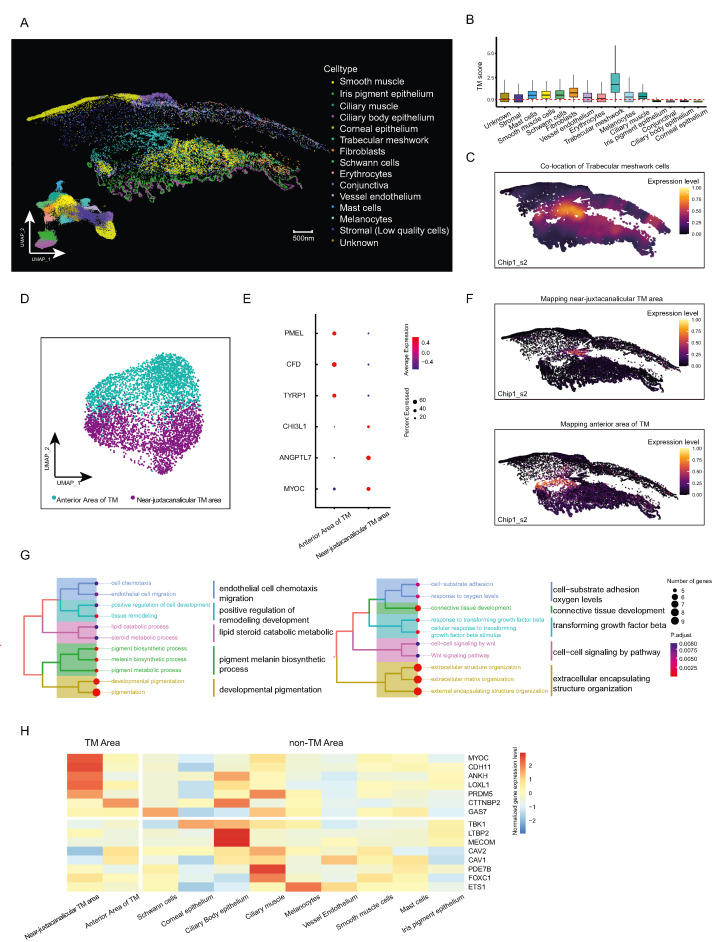
Elevated intraocular pressure (IOP) is recognized as a significant contributor to the development of various ocular diseases, especially glaucoma. The delicate balance between the generation and drainage of aqueous humor (AH) in the anterior chamber angle regulates the real IOP level. Despite extensive research, our understanding of the cellular components, tissue structure, and functional heterogeneity within the AH circulatory system (AHCS) remains incomplete, hindering the progression of effective and accessible treatment strategies for IOP intervention. Therefore, the state-of-the-art spatiotemporal single-cell omics stands poised to furnish an invaluable spatial cellular map of the AHCS, thereby laying the foundation for subsequent in-depth investigations. Meanwhile, this innovative approach promises to offer novel insights into the pathogenesis and management of IOP regulation. Here, we utilized nanoscale resolution-spatial enhanced resolution omics-sequencing (Stereo-seq1) to obtain in situ gene expression profiles of AHCS in human eyes (Fig. S1A). We generated two optimal cutting temperature compound-embedded chips of the trabecular meshwork (TM) and surrounding tissue from four samples (Table S1), which were later cut into layers of 10-μm-thickness cryosections for Stereo-seq and hematoxylin-eosin staining. Considering that transcript capture was performed at a subcellular level using a DNA nanoball sequencing technique, we integrated a semi-automated spatial omics methodology with cell segmentation using the GEM3D-toolkit (https://github.com/BGI-Qingdao/GEM3D_toolkit) to acquire a single-cell resolved transcript of AHCS in spatial scenarios. This method allowed us to partition two Stereo-seq chips into 60,638 putative single cells by assigning transcripts to each defined cell area at single-cell resolution (detailed in supplementary methods and materials and Fig. S1B). After the following quality control, we finally detected 978.5 DNA nanoball spots per cell area on average, with the per-cell detection of 885.4 unique molecular identifiers and 445.3 genes. Chip1 and Chip2 were composed of 32,081 and 28,557 cells, respectively, covering 24,775 genes (Fig. S1C and Table S2).