Clinical and genetic heterogeneity of adult polyglucosan body disease caused by GBE1 biallelic mutations in China
Clinical and genetic heterogeneity of adult polyglucosan body disease caused by GBE1 biallelic mutations in China
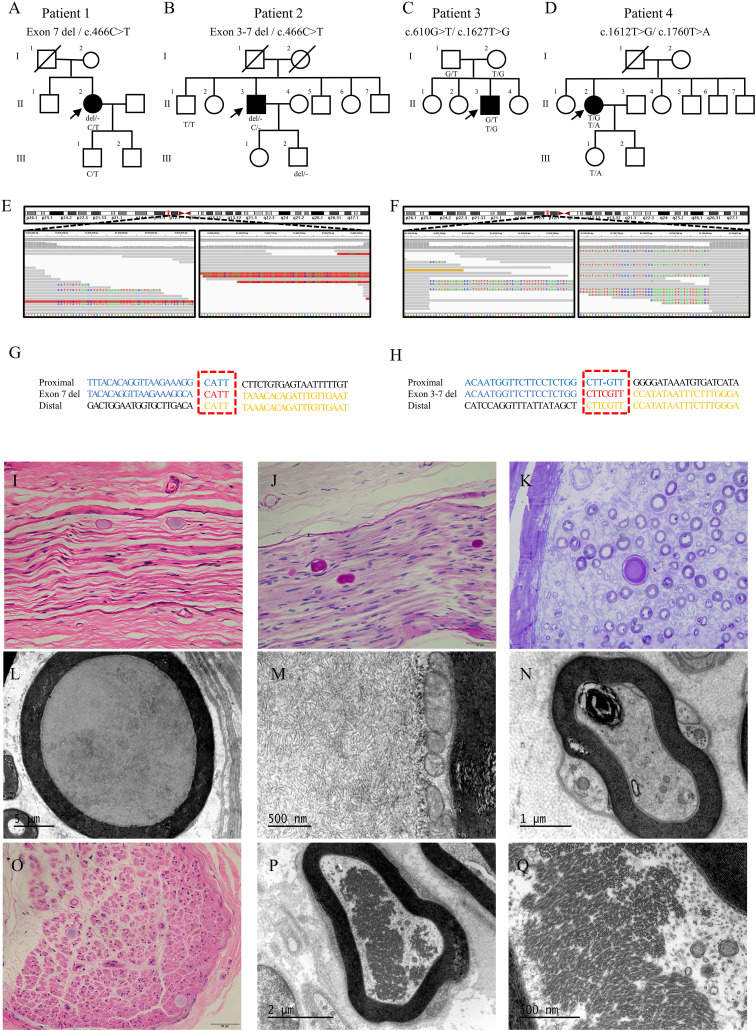
Adult polyglucosan body disease (APBD) is a rare and highly heterogeneous glycogen storage disorder due to biallelic variants in GBE1.1 Typical APBD presentations include gait abnormalities with polyneuropathy, leukodystrophy, neurogenic bladder, and mild cognitive impairment. Differential diagnosis of APBD encompasses a large spectrum of conditions including axonal and demyelinating sensorimotor polyneuropathy, progressive spastic paraparesis, and leukodystrophies. The majority of APBD patients are Ashkenazi-Jewish harboring a homozygous GBE1 mutation, c.986A > C. There have been a few APBD patients reported in East Asia. In addition, the large deletion mutations of GBE1 have only been previously described in early congenital neuromuscular patients.2 In this study, we depicted the phenotypic heterogeneity in four APBD patients in China and identified five single nucleotide variants and two large deletion variants in GBE1. These findings expand the clinical and genetic spectrum of APBD patients and reaffirm the importance of a more comprehensive and cautious diagnostic approach.