Identification of respiratory chain complex I deficiency due to NDUFA5 variants as a novel cause of infantile fatal disease
Identification of respiratory chain complex I deficiency due to NDUFA5 variants as a novel cause of infantile fatal disease
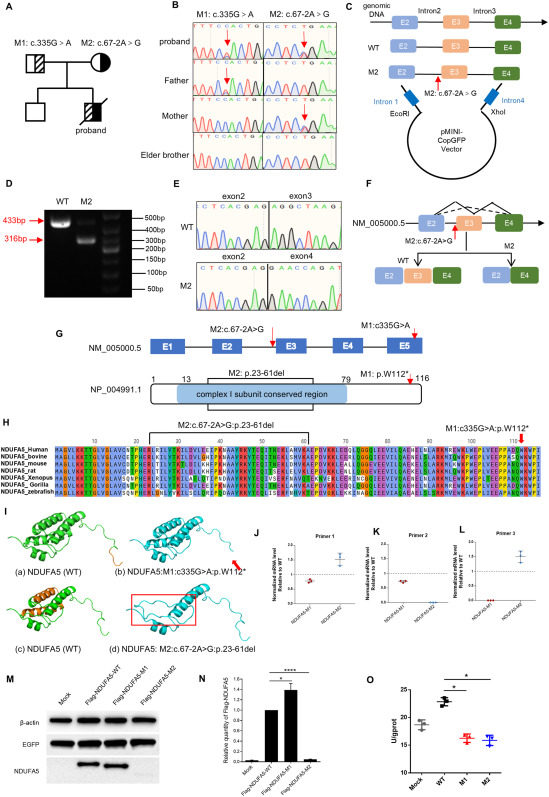
Mitochondrial complex I (CI), also known as NADH-ubiquinone oxidoreductase, represents the largest and most intricate component of the mitochondrial oxidative phosphorylation (OXPHOS) system, which is composed of 44 different subunits in humans and is assembled from the 14 core subunits.1 CI, a key component of the electron transport chain, represents a crucial site for the physiological production of reactive oxygen species (ROS) during cellular respiration, commonly recognized as being intimately linked to the oxidative damage that occurs subsequent to hypoxic exposure.2 Complex I is a distinctive boot-shaped structure composed of 14 core protein subunits organized into two main domains. The N module, located at the top of the matrix arm, assumes a pivotal role in catalyzing the oxidation of NADH. Meanwhile, the Q module functions as a vital connector between the substrate arm and the membrane arm, facilitating the transfer of electrons. The Q module comprises a set of proteins, including NDUFS2, NDUFS3, NDUFS7, NDUFS8, NDUFA9, and notably NDUFA5, which serves as a nuclear-encoded structural accessory subunit positioned within the peripheral segment of Complex I’s Q model. Previous studies have conclusively demonstrated that the lack of the NDUFA5 subunit leads to the loss of the membrane arm subcomplex. One of the interacting subunits of NDUFA5 is NDUFS2, which is a core subunit of mitochondrial complex I and is encoded by nuclear DNA. NDUFS2 plays a crucial role in the catalytic activity and assembly of mitochondrial complex I.3,4 Mitochondrial diseases are a group of inherited metabolic disorders with a high degree of clinical and genetic heterogeneity.5 Epidemiological data revealed that CI deficiency carries a grave prognosis, with approximately 50% of affected patients succumbing within the initial two years of life, while merely a quarter manage to reach 10 years old. Lactic acidosis is a common feature of the disease caused by CI variants and is accompanied by other symptoms, such as cardiomyopathy or leukodystrophy. In this study, we described a four-month-old child with progressive neurological deficits, postpartum lactic acidosis, encephalopathy, developmental abnormalities, metabolic abnormalities, and respiratory failure. Whole genome sequencing (WGS) identified two heterozygous variants of the NDUFA5 gene. A series of in vitro functional experiments were conducted and demonstrated that the NDUFA5 gene was associated with CI functional defects for the first time.