Non-canonical role of “S6K1–SGK1” pathway in neuronal necroptosis following traumatic brain injury
Non-canonical role of “S6K1–SGK1” pathway in neuronal necroptosis following traumatic brain injury
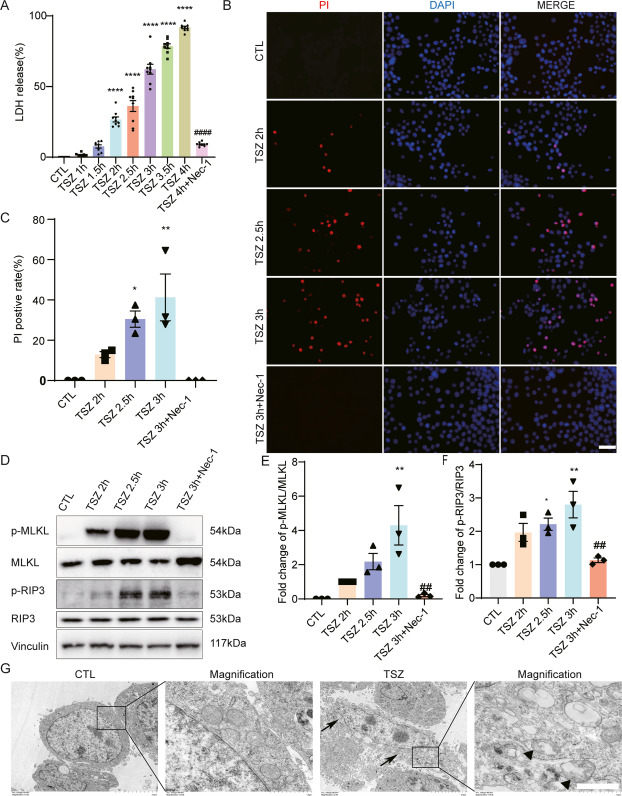
Traumatic brain injury (TBI) is characterized by high rates of death and disability. Necroptosis is reported to be involved in neuronal death after TBI. However, additional molecules and related mechanisms underlying necroptosis, particularly during TBI, remain to be elucidated. mTOR and two of its three substrates (4EBP1 and ULK1) are involved in necroptosis. However, direct evidence linking necroptosis to S6K, another key substrate of mTORC1, has been lacking. In this study, we aimed to investigate the regulated role of “S6K1-glucocorticoid-inducible kinase-1 (SGK1)” pathway in neuronal necroptosis after TBI. We first showed that the “S6K1–SGK1” pathway was activated during neuronal necroptosis in TNF-α/Smac mimics/Z-VAD-FMK-induced necroptotic cell model and mouse TBI model. Then, inhibition of the “S6K1–SGK1” pathway could decrease necroptosis by regulating the MLKL activation. Next, a rescue assay indicated that S6K1 may regulate necroptosis through modulating SGK1 expression, while not through binding with SGK1. Finally, S6K1 inhibition alleviated neuronal necroptosis, neuro-inflammation, and functional damage via SGK1 in mice after TBI. Our results showed a non-canonical role of “S6K1–SGK1” pathway in neuronal necroptosis following TBI in mice, which will provide a potential therapeutic target for necroptosis treatment in TBI and other necroptosis-related disorders.